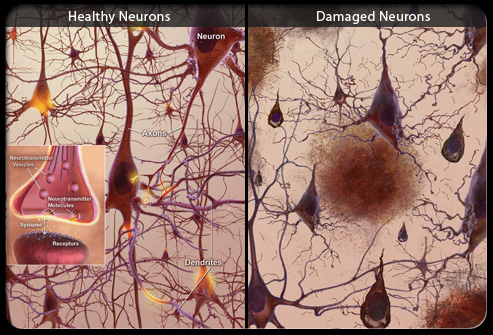
Le malattie da prioni sono un gruppo di malattie rare trasmissibili, che colpiscono l'uomo e gli animali, che si manifestano con sintomi neurologici progressivi e debilitanti, conseguenti ad alterazioni spongiformi del sistema nervoso centrale.
Queste malattie sono caratterizzate dall'accumulo di una proteina prionica anomala nel sistema nervoso centrale.
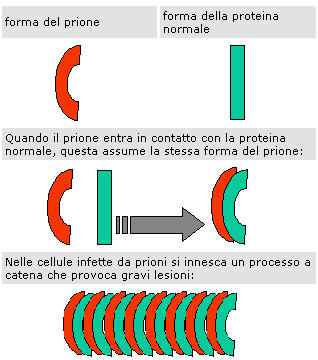
Il decorso delle patologie prioniche è sempre letale. La modalità di infezione è data da una particolare catena proteica ripiegata in maniera scorretta, che induce altre proteine ad assumere la stessa conformazione anomala, a loro volta di infettare le proteine adiacenti.
La malattia di Creutzfeldt-Jakob sporadica (CJD) è la forma più frequente e rappresenta circa l'85% delle malattie da prioni.
Le altre forme (5-15%) sono genetiche: CJD ereditaria, insonnia familiare fatale (FFI), malattia di Gerstmann-Sträussler-Scheinker e malattia familiare da prioni Alzheimer-simile.
Le forme acquisite (< 5%) comprendono il kuru, la CJD iatrogena e la nuova variante CJD (vCDJ).
Eziopatogenesi
Una volta ancorata ai neuroni, se viene a contatto con PrPsc, la PrPc subisce un cambiamento conformazionale (la differenza tra le due è solo conformazionale ed è legata alla mutazione puntiforme di un solo amminoacido). La PrPsc è poco solubile e quindi precipita formando aggregati insolubili chiamati placche amiloidi che innescano la reazione a catena.
Secondo il modello più accreditato, proposto dallo stesso Prusiner, la PrPc diviene pericolosa in seguito a un mutamento conformazionale, indotto da un prione infettante o da una mutazione genetica spontanea, che la trasforma in PrPres, la quale a sua volta agisce su altre PrPc con una reazione a catena.
Note
Voci correlate
- Malattia infettiva
- Prione